Table Of Content

Fingerprint based similarity clustering was then conducted to maximize the chemical diversity of the ranked compounds to be selected for bioassay testing. The best hit compound was then subject to a similarity search for chemically similar analogs and more inhibitory compounds were identified. Such identified non-β-lactam-based β-lactamase inhibitors have the potential to be used in combination therapy with lactam-based antibiotics against multi-drug resistant clinical isolates. With the steady rise in the number of confirmed positive and death cases from SARS-CoV-2 infection, computer-aided drug design (CADD) emerges as a fast and reliable technique in pharmaceutical and medicinal research since it not only saves time but also helps to cut costs of designing therapeutic agents [164]. Further, realizing the severity of COVID-19 and the lack of approved therapeutic agents warrants the need for finding potent drugs in less time, and the CADD method makes this possible by facilitating the discovery of new drugs or repurposing FDA-approved drugs whose safety and adverse effects are already known [165].
2 Site Identification by Ligand Competitive Saturation (SILCS)
Future-Proofing Cybersecurity in Drug Discovery - Genetic Engineering & Biotechnology News
Future-Proofing Cybersecurity in Drug Discovery.
Posted: Thu, 11 Jan 2024 08:00:00 GMT [source]
Numerous groups crowdsourcing the follow-up computational design and screening of merged and growing fragments helped to discover several SAR series, including a non-covalent Mpro inhibitor with an enzymatic IC50 of 21 μM. Further optimization by both structure-based and AI-driven computational approaches, which used more than 10 million MADE Enamine building blocks, led to the discovery of preclinical candidates with cell-based IC50 in the approximately 100-nM range, approaching the potency of nirmatrelvir65. The enormous scale, urgency and complexity of this Moonshot effort with more than 2,400 compounds synthesized on demand and measured in more than 10,000 assays are unprecedented and this highlights the challenges of de novo design of non-covalent inhibitors of Mpro. The most efficient and powerful process which governs entire drug discovery is structure-based drug designing (SBDD). The information about the target of small molecules, genetic information with their sequences, binding information, cytotoxicity, absorption, metabolism, excretion(ADMET) data, and other important biological information serve as the most efficient sources for accelerating the drug discovery process.
My comments on ICMJE Data-sharing proposal
Although the impacts of the recent structural revolution17 and computing hardware in drug discovery28 are comprehensively reviewed elsewhere, here we focus on the ongoing expansion of accessible drug-like chemical spaces as well as current developments in computational methods for ligand discovery and optimization. We detail how emerging computational tools applied in gigaspace can facilitate the cost-effective discovery of hundreds or even thousands of highly diverse, potent, target-selective and drug-like ligands for a desired target, and put them in the context of experimental approaches (Table 1). Although the full impact of new computational technologies is only starting to affect clinical development, we suggest that their synergistic combination with experimental testing and validation in the drug discovery ecosystem can markedly improve its efficiency in producing better therapeutics.
Professional development
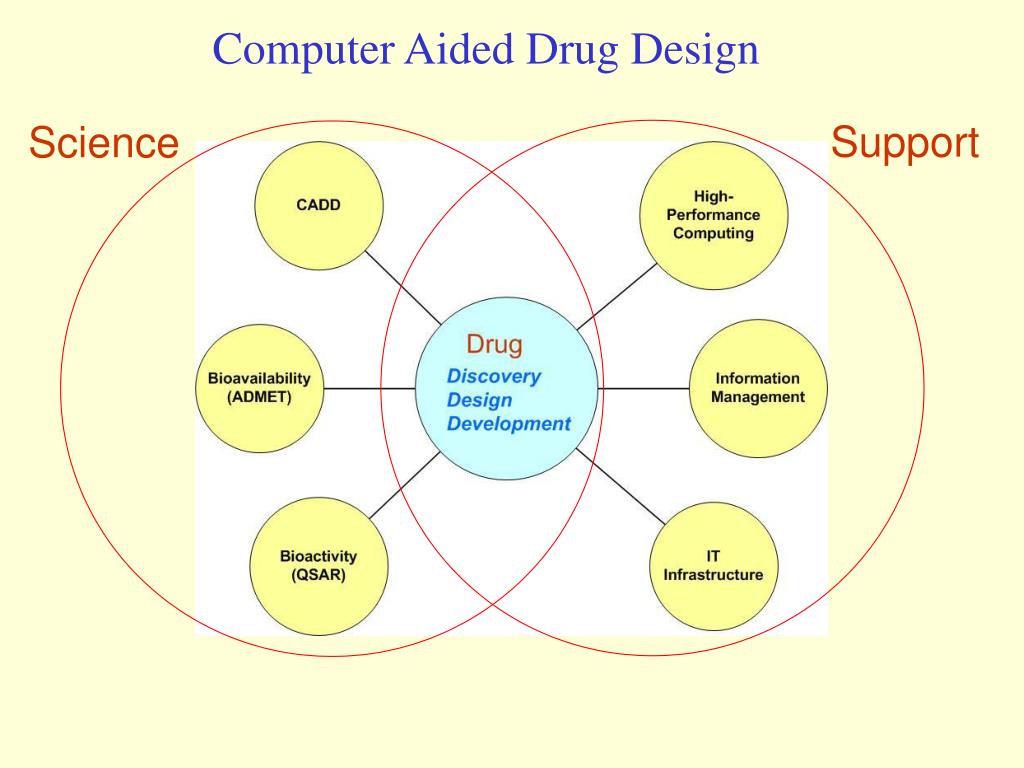
In the majority of the cases (40-60%), the drug failure at a later stage has been reported due to lack of optimum pharmacokinetic properties on absorption, distribution, metabolism, excretion, and toxicity (ADME/Tox) [5]. The use of computer-aided drug discovery (CADD) techniques in preliminary studies by leading pharmaceutical companies and research groups has helped to expedite the drug discovery and development process minimizing the costs and failures in the final stage [6]. The application of rational drug design as an integral part of CADD provides useful insights into the understanding of the binding affinity and molecular interaction between target protein and ligand. Additionally, lead identification in pharmaceutical research has been facilitated by the availability of supercomputing facility, parallel processing, and advanced programs, algorithms, and tools [7]. Furthermore, recent advancements in artificial intelligence (AI) and machine learning methods have greatly aided in analyzing, learning, and explaining the pharmaceutical-related big data in the drug discovery process [8].
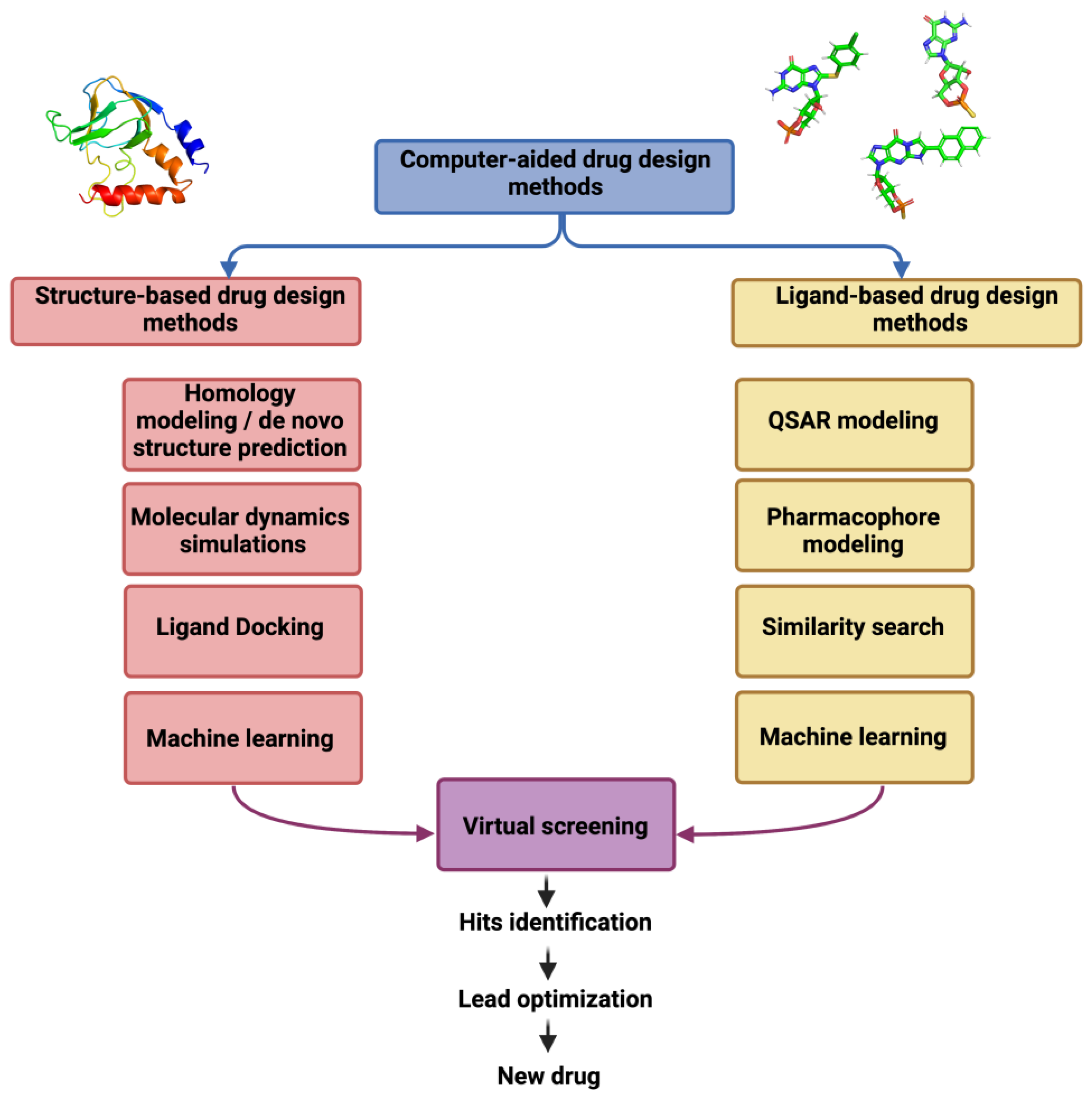
B. Ligand-based drug designing (LBDD)
Those hits, if any, are usually rather weak, non-selective, have suboptimal ADMET and PK properties and unknown binding mode, so their discovery entails years of painstaking trial-and-error optimization efforts to produce a lead molecule with satisfying potency and all the other requirements for preclinical development. Scaling of HTS to a few million compounds can be afforded only in big pharma, and it still does not make that much difference in terms of the quality of resulting hits. Likewise, virtual libraries that use in silico screening were traditionally limited to a collection of compounds available in stock from vendors, usually comprising fewer than 10 million unique compounds, therefore the scale advantage over HTS was marginal. Following the significant milestone that penicillin represents in human medical history, the battle between humans and bacteria has never settled down and becomes even more vigorous caused by the steady rise of drug resistance. This problem persists despite the availability of large number of antibiotic drugs, indicating the need for more novel antibiotic drug classes to overcome the resistance problem (1, 2).
Technology & Economic intelligence for Biopharmaceutical Strategy: toward a new approach
The bioactive conformation of the molecules within the target binding site can be incorporated into the pharmacophore model. Some frequently used programs which allow automatic construction of the pharmacophore model include Catalyst, PHASE, LigandScout, GALAHAD, and PharmMapper (Table 3). A good pharmacophore model also incorporates spatial constraints in regions occupied by inactive molecules and often optimized further to make the model less restrictive.
Pharmacophore models mapping
Though highly interrelated, identification of active scaffolds should be conceptually separated from drug design. Traditionally, the drug design process has focused on the molecular determinants of the interactions between the drug and its known or intended molecular target. Most antibiotics were designed to target proteins involved in intracellular processes, thus the outer membrane of bacteria needs to be penetrated for antibiotics to function. Drug resistance involving modifications of macromolecules in the outer membrane is a common issue that needs to be considered when searching for new antibiotics (122, 123).
5. Molecular Dynamic (MD) Simulation
The catalytic core of the enzyme resembles the human right hand with differentiated palm, fingers, and thumb domains. Targeting this enzyme to halt the viral replication seems an effective therapeutic approach since the active site of the RdRp is a highly conserved and accessible region [121]. Nsp15 is a uridine-specific endoribonuclease involved in RNA processing and widely distributed in all kingdoms of life. Its catalytic C-terminal domain exhibits sequence similarity and functionality of the EndoU family enzymes [122]. The active 234-kDa hexameric enzyme cleaves both single- and double-stranded RNA at uridine sites generating 2′,3′-cyclic phosphodiester and 5′-hydroxyl termini [123].
3. Artificial Intelligence and Drug Discovery
Our findings indicated that Ocotillone and Subsessiline have potential antileishmanial effects at pH 5 and 7, respectively, due to their high binding affinity to MOGS and interactions in the active center. This research indicates the promising anti-leishmanial activity of Ocotillone and Subsessiline, suggesting further validation through in vitro and in vivo experiments. The development of more robust generative chemical spaces can also be supported by new computational approaches in synthetic chemistry, for example, predictions of new iterative reaction sequences129 or synthetic routes and feasibility from DL-based retrosynthetic analysis130.
An ideal search algorithm should be fast and effective, and the scoring function must be capable of determining the physicochemical properties of molecules and the thermodynamics of interactions. At the early hit identification stage, the ultra-scale virtual screening approaches, both structure-based and AI-based, are becoming mainstream in providing fast and cost-effective entry points into drug discovery campaigns. At the hit-to-lead stage, the more elaborate potency prediction tools such as free energy perturbation and AI-based QSAR often guide rational optimization of ligand potency.
The bulky compounds which do not fit well within the binding site pocket are rejected during the lead identification procedure. To overcome limitations and improve accuracy in terms of predicting potent leads, regular updates of tools and algorithms are needed. Database reliability and high quality validated experimental molecules is to be developed and updated because many pharmacophores do not pass biological activity process due to non-availability of good quality data sets.
First, it can proportionally increase the number of potential hits in the initial screening31 (Fig. 2). This abundance of ligands in the library also increases the chances of identification of more potent or selective ligands, as well as ligands with better physicochemical properties. This has been demonstrated in ultra-large virtual screening campaigns for several targets, revealing highly potent ligands with affinities often in the mid-nanomolar to sub-nanomolar range20,21,22,23,26.
Here we present a docking protocol using the DOCK program (49) to illustrate the typical docking VS workflow. Boxes with solid lines indicate methods used in the CMY-10 study while boxes with dashed lines mark methods not used in the CMY-10 study but in other studies. Double arrows indicate the two techniques can be used interactively in several iterative drug design rounds. Although blind benchmarking and recent prospective screening success stories for the growing number of targets support utility of modern computational tools, there are whole classes of challenging targets, in which existing in silico screening approaches are not expected to fare very well by themselves.
Recently CADD methodologies have become a fundamental and indispensable tool in different junctures of drug discovery and development. Additionally, with the increasing availability and knowledge of biological/bio-macromolecule structures, as well as an exponential increase in computing power, it is now plausible to use these methods effectively without the significant loss of accuracy. CADD has also paved paths for the screening of selected compounds and synthesis of those entities for better therapeutics.








No comments:
Post a Comment